
FAQ
Nicking Loop™ enables highly efficient and accurate data copying (see question How the Nicking Loop™ copying is done?), ensuring reliable data propagation through rolling circle amplification (RCA) (see question How does the rolling circle amplification (RCA) work in Bridge Capture™ and Nicking Loop™?). This technique utilizes circular templates that are linearly amplified by Phi29 DNA polymerase, producing consistent and precise replication. Unlike PCR-based methods, RCA avoids issues such as uneven amplification, and data corruption by maintaining a linear amplification process. This approach provides a dependable solution for high-fidelity data copying and consistent data retrieval.
With Nicking Loop™ the specific retrieval/deletion sequences are embedded within the structure of library molecules. In a case of data deletion these sequences enable the selective deletion of predefined data or sequence subsets using highly specific quide- oligonucleotides. These oligonucleotides direct enzymatic activity to target and remove specific portions of the library or block the amplification of selected subsets of library molecules. This approach ensures precise data deletion while preserving the integrity of the remaining library content.
With Nicking Loop™, the specific query sequences/motifs are embedded within the structure of library molecules. These sequences enable the targeted retrieval of data or sequence subsets by employing highly specific query-primers.
These primers initiate selective rolling circle amplification (RCA) -based copying (see question How the Nicking Loop™ copying is done?), allowing specific amplification of the desired portions of the data library with high precision and efficiency.
DNA can act like a database by utilizing the molecular structure of DNA to store and retrieve information in a compact and durable format. In DNA, the natural sequences of the four DNA bases (A, T, C, G) are engineered to encode digital data, such as text, images, or even video files. These sequences serve as a biological equivalent to binary code in conventional data storage systems.
Information is encoded into synthetic DNA using chemical synthesis methods and retrieved through DNA sequencing technologies. Specific data can be queried from these DNA-based databases, functioning similarly to traditional digital databases in terms of data retrieval and deletion.
Synthetic DNA is incredibly dense as it can store up to a billion gigabytes in a single gram. It is also remarkably stable, capable of preserving data for thousands of years under the right conditions. This makes DNA a promising medium for long-term, high-density data storage.
Circular libraries are circular ssDNA molecules. A circular library is created in the process of converting a primary sample, such as cfDNA, genomic DNA, or an existing sequencing library, into a stable, circular single-stranded DNA (cssDNA) form. The conversion into circular library can be done either by targeting, using the Bridge Capture™ (see question How does GapFill work in the Bridge Capture™?), or by using the Nicking Loop™ conversion & indexing (see question How is the Nicking Loop™ conversion & indexing done?) for convert all the sample DNA. This transformation preserves the integrity of the sample while enabling easy and efficient access to its entire content for downstream applications. This process ensures reliable replication with minimal bias, enabling consistent and accurate analysis. Circular library also makes it possible to re-analyze and distribute rare or valuable DNA samples, preserving them for further research and broader use.
Bridge Capture™ and Nicking Loop™ enable the production of two distinct types of DNA libraries: linear and circular libraries.
Circular libraries are generated either from the primary gap-fill reaction of the Bridge Capture™ workflow (see question How does GapFill work in the Bridge Capture™?), converting any DNA into circular libraries (see question How is the Nicking Loop™ conversion & indexing done?) or through the Nicking Loop™ copying process (see question How the Nicking Loop™ copying is done?), which directly amplifies circular single-stranded DNA (ssDNA) molecules. These circular ssDNA molecules can function either as templates for additional amplification generations or as ready-to-use sequencing libraries. The size of the circular libraries typically ranges from 250 to 400 bases.
Linear libraries are produced by PCR amplification. Template for amplification is either a circular library or a concatemeric product generated by rolling circle amplification (RCA) (see question How does the rolling circle amplification (RCA) work in Bridge Capture™ and Nicking Loop™?). The resulting linear libraries typically range in size from 250 to 350 base pairs.
Yes.
The Nicking Loop™ conversion & indexing (see question How is the Nicking Loop™ conversion & indexing done?) enables the creation of circular single-stranded DNA (ssDNA) whole-genome libraries, facilitating comprehensive whole-genome sequencing.
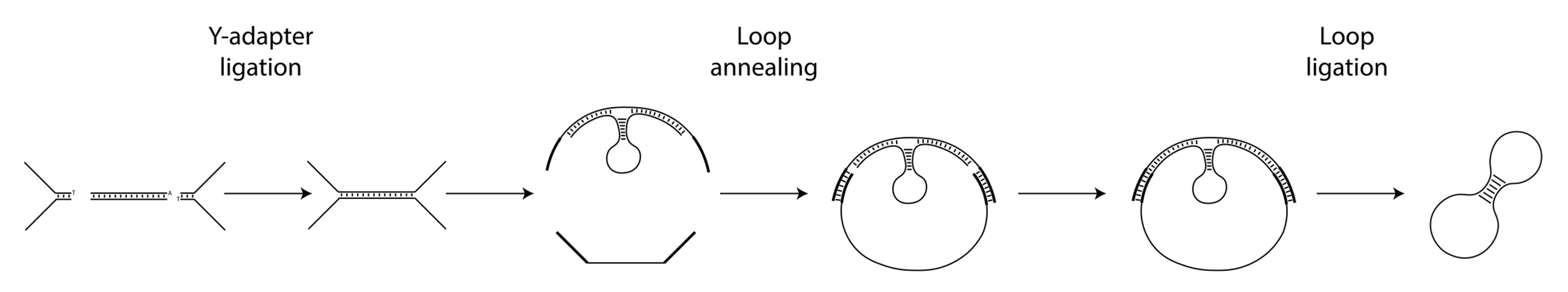
The Nicking Loop™-converted circular DNA molecule is constructed from a DNA template, a bridge oligomer, a Nicking Loop, and Y-adapters. The process involves the following steps:
1. Template Preparation: The template, which can be any unknown genomic DNA or linear library molecule, is ligated with Y-adapters at its ends to create binding sites for a universal bridge oligomer complex.
2. Circularization: The bridge oligomer complex binds to both Y-adapter ends of the template, forming a circular DNA structure. A Nicking Loop is also introduced here to the construct, enabling an early stage sample indexing.
3. Ligation: Nicks in the circular structure are sealed, completing the formation of a single-stranded DNA (ssDNA) circle. Excess non-circularized molecules are then removed using exonucleases.
4. Amplification: The intact circular molecules are amplified using rolling circle amplification (RCA).
For further details, refer to the question: How the Nicking Loop™ copying is done?
Yes, optionally.
Bridge Capture™ as well Nicking Loop are designed to be compatible with UMIs (see question What is a UMI and how are they implemented in the data processing?). In Bridge Capture™ UMIs are located in the probe arms (see question What is a probe?), whereas in Nicking Loop™ UMIs are incoporated to the loop structure (see question Can a sample be indexed with Nicking Loop™?). UMIs are useful for ultrasensitive quantitative applications, such as detecting low allele fractions from cfDNA. However, UMIs may not be necessary for the applications where simple presence or absence quantification is required.
In the Bridge Capture™, samples can be indexed with a Nicking Loop at the very beginning of the protocol; during the target capture reaction of Bridge Capture™ workflow (see question How does the targeting work in the Bridge Capture™?), in where probe complexes and indexed Nicking Loops, interact with the target material.
In the Nicking Loop™ conversion & indexing the Nicking Loop is introduced in the circularization step (see question How is the Nicking Loop™ conversion & indexing done?).
The early indexing facilitates the identification and differentiation of individual samples while enabling precise tracking of molecules throughout the downstream analysis.
Yes.
The loop structure of the Nicking Loop molecule is designed to accomodate pre-determined index and/or Unique Molecular Identifier (UMI) sequences. During the Bridge Capture™ target capture (see question How does the targeting work in the Bridge Capture™?) and gap fill reactions (see question How does GapFill work in the Bridge Capture™?) Nicking Loops are attached to the probe complexes. In the Nicking Loop™ conversion & indexing the Nicking Loop is introduced in the circularization step (see question How is the Nicking Loop™ conversion & indexing done?). Both workflows generate indexed circular ssDNA library molecules. The incorporation of these index sequences and UMIs ensures accurate sample identification and sensitive molecular tracking during downstream analysis.
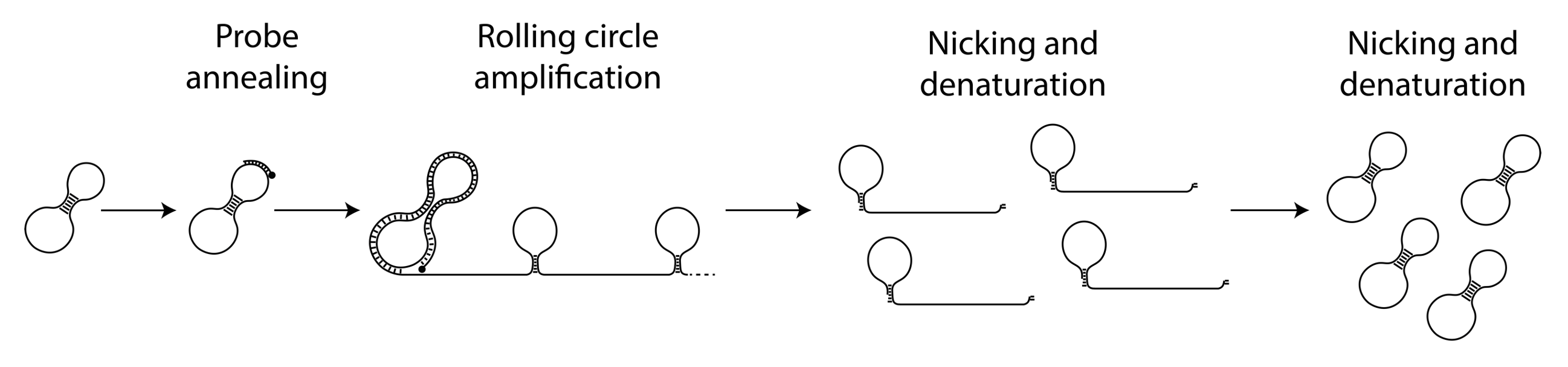
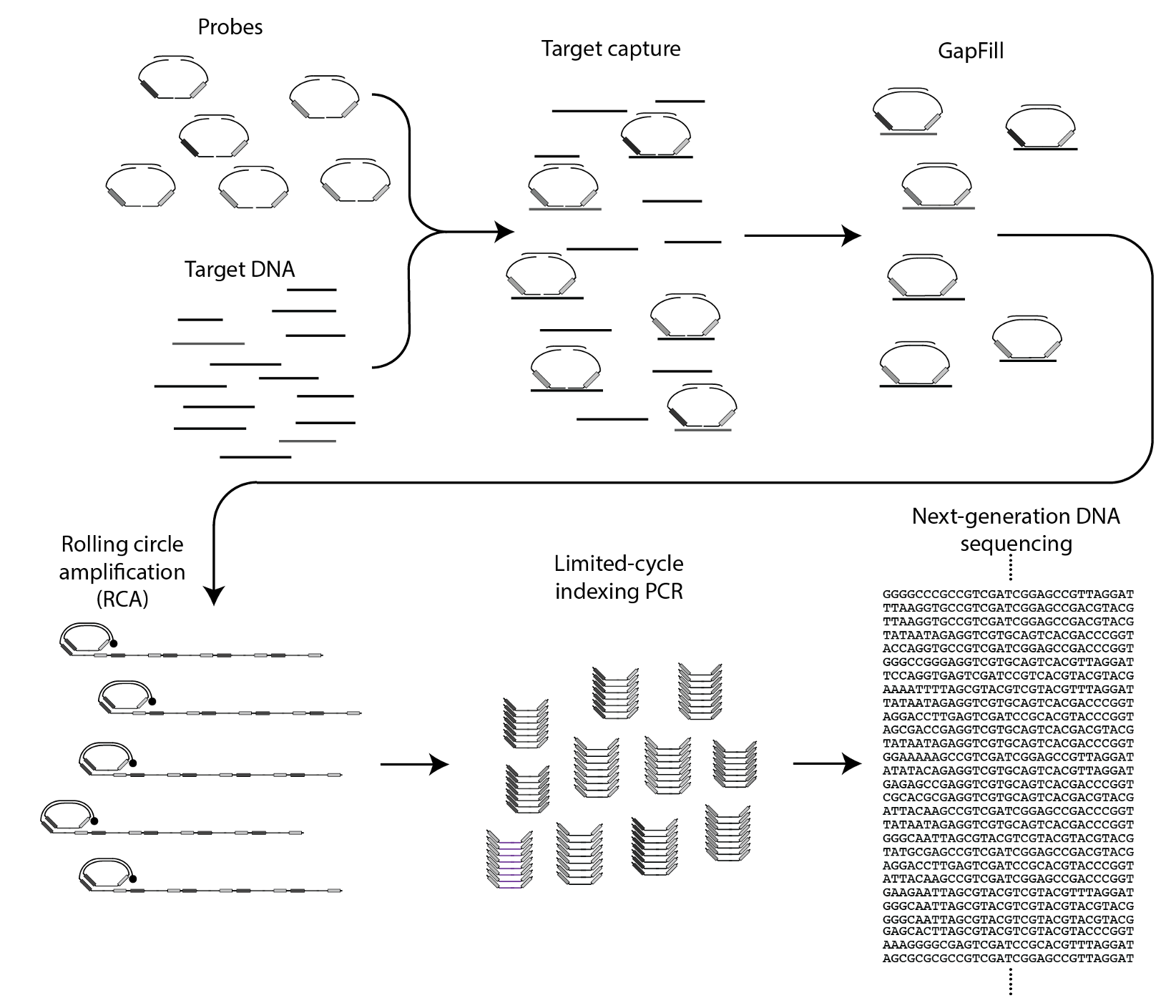
The circular ssDNA molecules containing the Nicking Loops are either achieved by targeting in the Bridge Capture™ (see How does GapFill work in the Bridge Capture™?) or with non-targeted Nicking Loop™ conversion & indexing (seequestion How is the Nicking Loop™ conversion & indexing done?). These serve as a template for the linear rolling circle amplification (RCA) reaction (Figure 1 step 1). RCA produces a long concatemeric molecule containing multiple adjacent linear copies of the original circular template (see question How does the rolling circle amplification (RCA) work in Bridge Capture™ and Nicking Loop™?).
A long linear molecule generated by RCA contains sequence structures that facilitate the formation of local hairpin loops at fixed positions (Figure 1 step 2). Each segment, spanning from the complete loop structure to the start of the next adjacent loop, represents one full cycle around the circular template. The double-stranded stem of each loop includes a recognition site for a specific nicking endonuclease enzyme, able to cleave only one strand of the stem sequence. This targeted digestion acombanied with mild heating cuts the long concatemeric molecule into multiple pre-defined DNA fragments, each representing a linear copy of the original circular template (Figure 1 step 3). The ends of these fragments contain complementary sequence motifs, enabling them to self-anneal. With the addition of a ligase, the fragments can be re-circularized into complementary copies of the original template (Figure 1 step 4). These circular molecules can serve either as templates for further amplification cycles or as ready-to-use sequencing libraries with circular sequencing platforms (see What are the outputs of Bridge Capture™ and Nicking Loop™ processes?).
UMIs are sequences associated with each read that permit identifying the original molecule from which amplification took off. UMIs permit accurate target quantification as well as error correction. UMIs are introduced by incorporating random nucleotides into predefined regions of the probes. When the probes are sequenced, the UMI sequences are also read. The UMIs are designed to be long enough to create sufficient variation, so that each probe hybridization event to the target material will have a high probability of having its own unique UMI. This high level of variation is crucial for accurate quantification, as it ensures that each individual molecule is accurately identified and counted. This permits even rare variants to be quantified with high accuracy.
The UMI sequences are used in the Bridge Capture™ and Nicking Loop™ data processing in error correction. Error correction assumes that a single UMI should be associated with a single sequence, and any deviation from this is a result of an amplification or sequencing error. Taken together with the read count, the UMI sequences permit very accurate quantification of molecular counts with a high signal-to-noise ratio.
Bridge Capture™ is a patented laboratory process workflow. The bridge structure connects the two target-specific probes, enabling the capture of the target region and its subsequent linear amplification. This innovative design allows the targeted region to be copied to the probe gap. This permits capture of relatively long target regions while keeping the cost of probe synthesis down by using shorter individual oligos. Bridge is the turquoise structure in Figure 1.
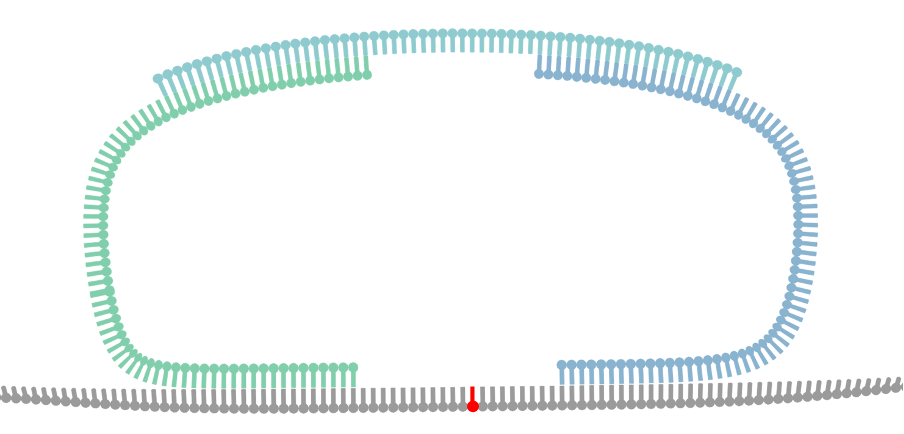
The probe-bridge constructs are introduced into a hybridization reaction with the sample material. The number of probe-bridge constructs is significantly higher than the sample material to improve the kinetics of the hybridization. Optionally, a Nicking Loop can be introduced for early sample indexing and Nicking Loop™ functionalities. From here the workflow advances to the gapfill reaction (see question How does GapFill work in the Bridge Capture™?).
In the Bridge Capture™ workflow, the process of circularizing the probe involves several steps. After the targeting (see question How does the targeting work in the Bridge Capture™?) a polymerase is introduced to fill the gap between the two probes. The polymerase uses the target sequence as a template to fill in the gap between the probes. As the polymerase extends the sequence, it eventually meets the other probe, and a ligase joins the two probes together. Here the optional Nicking Loop is incorporated to the structure as well.
Furthermore, during the polymerase and ligase reaction, the gap that exists between the probes in the bridge area is also filled, thus ligating the probes together from this end as well. This leads to the formation of a full circular molecule with the bridge still hybridized to it. The bridge sequence is an essential part of the probe design, as it helps to hold the two probes together and maintain the circularized structure of the molecule.
The resulting circularized probe, a circular library (see question What is a circular library?), is now ready for amplification through RCA (see question How does the rolling circle amplification (RCA) work in Bridge Capture™ and Nicking Loop™?) or it can be optionally sequenced directly with a circular-based sequencer like Complete Genomic’s DNBSEQ, Element’s Aviti or PacBio’s Onso.
Rolling circle amplification (RCA) is a nucleic acid amplification technique that involves the circularization of an ssDNA template, followed by repeated extension of a DNA polymerase around the circularized molecule, resulting in the formation of long, branched DNA molecules.
In Bridge Capture™ and Nicking Loop™ processes the circular template is either a Bridge Capture™’s GapFill product (see question How does GapFill work in the Bridge Capture™?) or the Nicking Loop™’s conversion & indexing product (see question How is the Nicking Loop™ conversion & indexing done?). In both cases the RCA is primed either from the bridge oligo or from a separate primer, which is extended by Phi29 DNA polymerase. As the Phi29 polymerase progresses around the circularized template, it displaces the previously synthesized strand, resulting in the formation of a long, concatemeric DNA molecule. Phi29 is highly processive, and extension results in the formation of many copies of the original circular template, leading to efficient linear amplification of the target sequence.
The resulting concatemeric ssDNA can be processed further for linear sequencing platforms with limited-cycle indexing PCR (see question How does the limited-cycle indexing PCR work in the Bridge Capture™?. The concatemeric ssDNA product can also be sequenced with long-read sequencing platforms, such as Oxford Nanopore Technologies, or the concatemers can be nicked and folded back into circular libraries (see question How the Nicking Loop™ copying is done?).
Limited-cycle indexing PCR uses primers with overhangs to introduce the required adapters for the sequencing platform of choice. The PCR is done in limited cycles to minimize the exponential amplification as the main amplification is done in RCA.
Yes.
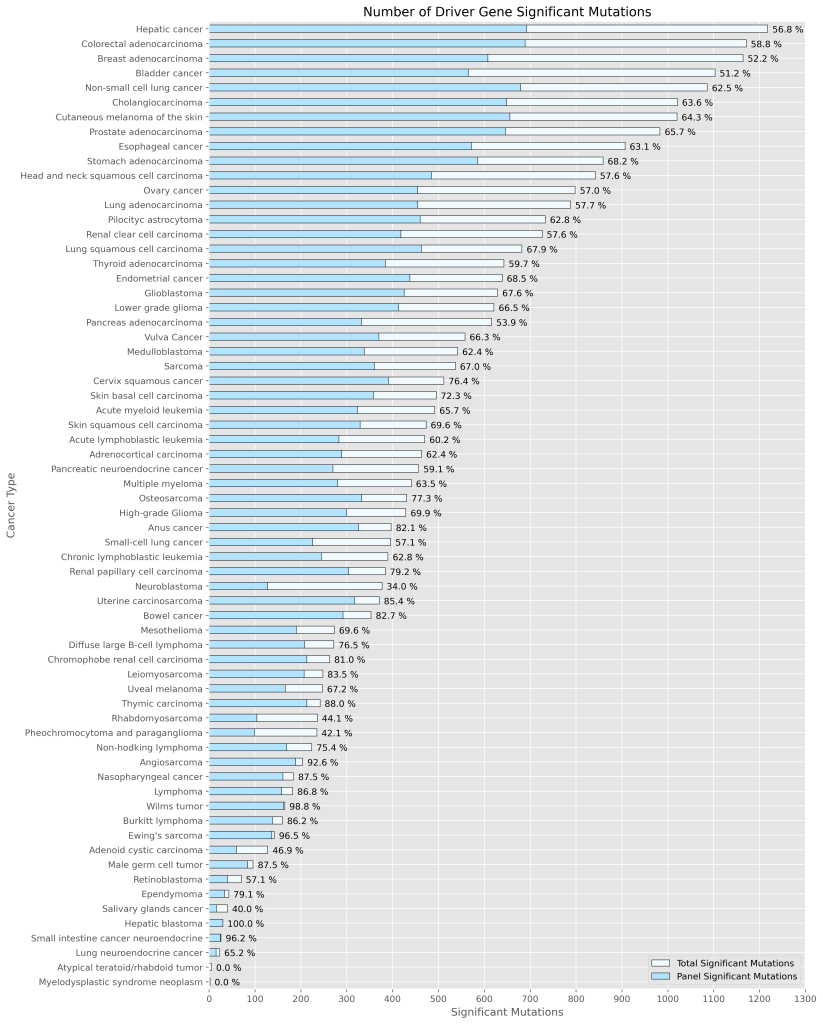
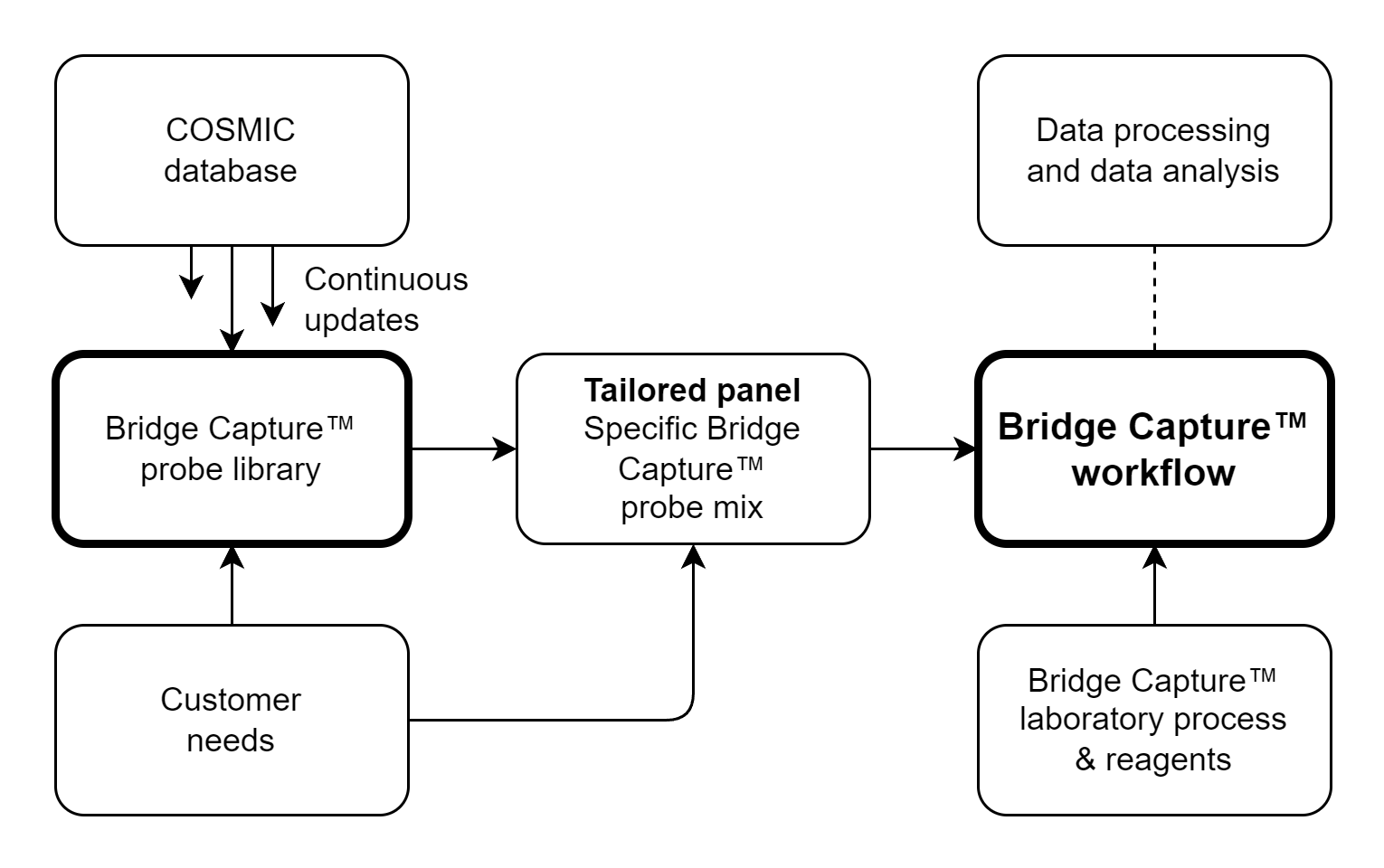
Genomill has developed a panel with 887 probes which targets 123 genes containing 24,236 mutations, from which 1,870 are oncologically relevant mutations (see Figure 1). The panel design is based on the COSMIC database, CMC v.99 (https://cancer.sanger.ac.uk/cosmic).
Genomill’s panel is fully modular, since panel can be split into multiple smaller panels and from these the desired subpanels can be combined back into one bigger panel, still retaining their probe performance.
Bridge Capture™ process is illustrated in Figure 1 and the technology is illustrated in detail in Figure 2.
Bridge Capture™ is an efficient targeted pre-sequencing technology which offers a rapid, highly sensitive, platform agnostic and cost-effective molecular quantification solution.
In Bridge Capture™ the sample is targeted with Bridge Capture™ probes (see question How does the targeting work in the Bridge Capture™?) are included in the probes.
Once the probes have found their intended targets, the gap between probes is copied from the target sequence and ligated to form gap-filled circular probe construct (see question How does GapFill work in the Bridge Capture™?), a circular library, which can be optionally sequenced directly with a circular-based sequencer like Complete Genomic’s DNBSEQ, Element’s Aviti or PacBio’s Onso. This circular library is linearly amplified by RCA to generate multiple copies of the circular probe (see questions How does the rolling circle amplification (RCA) work in Bridge Capture™ and Nicking Loop™?), which can be optionally sequenced with long-read sequencing platforms, such as Oxford Nanopore Technologies.
After linear amplification, sequencing platform-specific adapters are introduced through a limited-cycle indexing PCR (see question How does the limited-cycle indexing PCR work in the Bridge Capture™?). To ensure optimal quality and purity, the resulting libraries are purified with bead purification protocols. This step helps to remove any impurities and contaminants that may affect the accuracy of the sequencing data.
No.
Genomill obtains probe oligomers and other oligonucleotides from well-established commercial providers.
No.
Genomill has developed an efficient and highly scalable probe design method. It is based on a streamlined proprietary in-house algorithm able to design functional probes in minutes. GenomillÕs probe production pipeline involves also rigorous experimental testing, ensuring the highest possible quality of each probe used.
Not necessarily.
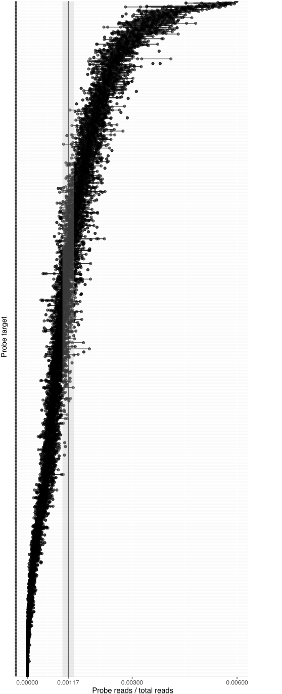
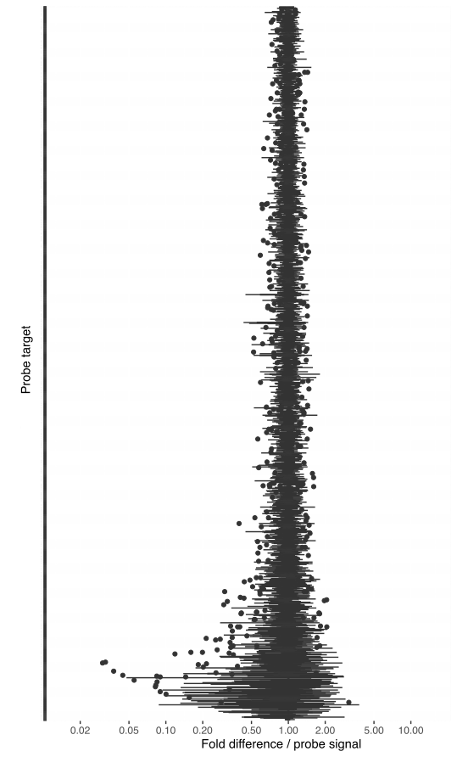
With our in-house probe designer, the probes typically work with good evenness (see Figure 1). We have shown that panel evenness can be further improved by adjusting individual probe concentrations.
The standard gap length in Bridge Capture™ is approximately 50 base pairs. This length was chosen to strike a balance between the small size of cfDNA fragments and the number of mutations that can be captured by a single probe pair when the gap length is increased. The longest gap length tested so far is 110 base pairs, while the shortest gap tested was a nick between the probes, which was used to quantify specific molecules.
Probe arms are 20 base pairs long on average. This results in a typical combined footprint of 90 bp for left and right probes together with the gap region.
There is no predefined upper limit for the targets (number of probes) in a panel. So far, our largest panel has contained 887 probes and it worked as expected.
Probe design requires detailed information of target DNA sequence or genomic coordinates of the mutation.
Yes.
New probes can be introduced into an existing panel when required. Experimental panel validation is mandatory after introduction of new content.
Probe mix is a solution containing specific probes, or a physical embodiment of a panel it represents. Probe mix is an essential part of the Bridge Capture™ chemistry.
Yes.
The Bridge Capture™ and Nicking Loop™ have been designed to produce sequencing libraries that are compatible with any NGS platform. This is made possible by the circular libraries (see question How is the Nicking Loop™ conversion & indexing done?) which can be sequenced either with short-read or long-read platforms. See question What are the outputs of Bridge Capture™ and Nicking Loop™ processes? for an overview of how the Bridge Capture™ and Nicking Loop™ technology outputs fit to different sequencing platforms.
Yes.
Bridge Capture™ can analyze variety of the different sample materials. Routinely tested sample types are cfDNA, gDNA, and cDNA. The technology has demonstrated promising results on direct targeting of RNA, as well as capturing unpurified DNA from urine and saliva.
With Nicking Loop™ we are currently validating the dynamic range of the method.
Yes.
The smallest amount of patient cfDNA tested with Bridge Capture™ thus far has been approximately 2000 genomic copies (5 ng). The detection of low allelic fractions benefits from a larger amount of sample used in the workflow.
With Nicking Loop™ we have not conducted large enough studies on this topic yet.
No.
After the cfDNA is extracted from the sample, purified material can be used straight for the Bridge Capture™ and Nicking Loop™ workflows. Sample doesnÕt need to be diluted, as it is unlikely that amount of cfDNA in sample would saturate the number of probes used in the Bridge Capture™ or the adapters in the Nicking Loop™.
Yes.
Bridge Capture™ workflow permits sample pooling after the GapFill (see question How does GapFill work in the Bridge Capture™?) without the need for normalization whereas other commercially available technologies usually pool the pre-indexed amplified libraries just before sequencing. This approach ensures that as the number of samples processed in parallel increases, the associated increase in workflow duration, cost, and complexity does not follow a linear trend, offering a distinct advantage in efficiency and scalability. Our pooling approach is currently protected within our patents and it is undergoing further improvements.
When using Nicking Loops, the samples can be pooled after introducing and ligating the Nicking Loops. This would be after GapFill when using Nicking Loops in Bridge Capture™ (see question How does GapFill work in the Bridge Capture™?). In case of Nicking Loop™ conversion and indexing the pooling can be done after the circularization step (see question How is the Nicking Loop™ conversion & indexing done?).
Yes.
Bridge Capture™ has been demonstrated to reliably detect SNVs, indels, and gene fusions. We also have preliminary results of detecting CNVs. Bridge Capture¨ can detect mutations associated with any cancer type, given the right panel design.
Nicking Loop™ has been demonstrated with SNVs and due to the shared functions with Bridge Capture™ we assume Nicking Loop™ to handle the same mutation types.
A probe is a DNA oligomer that binds to complementary DNA sequence. Bridge Capture™ uses probe pairs, or probe arms, which are bound together by a third, non-target-binding oligomer. See figure 1 (probes in green and blue). The first enzyme reaction called GapFill takes place after the probe hybridizes to its target (see FAQ interlinks below).
The probe library contains all the probes Genomill has developed.
A panel is a subset of the probe library.
In context of cancer diagnostics, a panel is a predefined set of probes to target specific regions of the genome to detect genetic mutations, variations or other alterations associated with a particular cancer type.
Bridge Capture™ provides various advantages when compared to different liquid biopsy techniques. We therefore provide three separate answers for this question where Bridge Capture™ is compared to amplicon-based techniques (see question Does Bridge Capture™ have advantages over amplicon-based techniques?), hybrid capture-based techniques (see question Does Bridge Capture™ have advantages over hybrid capture techniques?) and molecular inversion probe-based techniques (see question Does Bridge Capture™ have advantages over Molecular Inversion Probes?).
One key benefit Bridge Capture™ has over all of these techniques is the capability to pool the samples directly after the targeting step without quantification. This approach ensures that the workload does not increase linearly with the number of samples, offering a distinct advantage in scalability (see question Can samples be pooled in Bridge Capture™ or Nicking Loop™ workflows?).
Bridge Capture™ is a highly sensitive molecular quantification technique. Its typical oncological use cases involve quantification of mutations (including SNVs, indels and gene fusions) and CNVs (see question Can Bridge Capture™ and Nicking Loop™ detect different mutation types?). One use case is mutation/CNV detection from tumor DNA. Another use case is mutation/CNV detection from cfDNA extracted from patientÕs blood plasma, which has applications in Tx and MRD.
Yes.
Amplicon-based techniques are based on multiplexed PCR which has inherent limitations in multiplexing, sequencing evenness and rare target detection. Amplicon-based workflows can be improved by introducing UMIs but this leads to a more complicated workflow.
Bridge Capture™ overcomes these limitations.
Bridge Capture™ provides improvements in scalability, sensitivity and workflow simplicity. Bridge Capture™ is a simple protocol, optionally utilizing UMIs (see question Does Bridge Capture™ and Nicking Loop™ use UMIs?), with infinite scalability, high evenness, and high sensitivity for rare variants. This results in improved performance as well as a streamlined, highly cost-efficient protocol.
Yes.
Bridge Capture™ provides improvements in chemistry and workflow costs (see question What’s the estimated cost of Bridge Capture™ and how is it determined?), as well as improvements in speed, and simplicity. The Bridge Capture™ workflow itself is simple and cost-efficient and results in a sequencing library with high target evenness. Since this library is focused on pre-selected loci, even very rare variants can be called with moderate sequencing depth.
In contrast, hybrid capture-based techniques are based on sequencing library preparation workflows and extend them by introducing an additional enrichment step where the desired parts of the sequencing libraries are enriched using capture probes. Hybrid capture techniques permit profiling a large number of mutations from sample material but suffer from expensive and complicated laboratory workflows, sequencing unevenness, high sequencing requirement, and complicated data analysis.
Bridge Capture™ overcomes these limitations and provides a lean, sensitive and cost-efficient alternative to hybrid capture.
Yes.
Bridge Capture™ has advantages in performance, scalability and cost-efficiency in comparison to molecular inversion probes.
Molecular inversion probes are composed of a lengthy single-stranded DNA molecules including linker region and target-specific segments at both ends. This continuous structure involves high probe synthesis costs as well as significant challenges and limitations on the probe design process. Molecular inversion probes are typically amplified using PCR primers specific to the linker sequence, resulting in uneven target amplification and probe cross-reactivity.
In contrast, Bridge Capture™ uses a design featuring short, target-specific probe arms connected by a bridge oligonucleotide (see question How does the targeting work in the Bridge Capture™?). This innovative structure significantly lowers the probe synthesis costs, streamlining the design and testing procedures. The modular nature of Bridge Capture™ – with each probe comprising three separate oligonucleotides – facilitates seamless implementation of novel design ideas and modifications on targeting parts, permitting approaches like sample barcoding on targeting, and PCR-free DNA sequencing libraries.
Unlike the conventional PCR amplification of molecular inversion probes using primers specific to the linker sequence, Bridge Capture™ utilizes linear signal amplification. This approach avoids probe cross-reactivity, ensuring highly uniform and reproducible results.
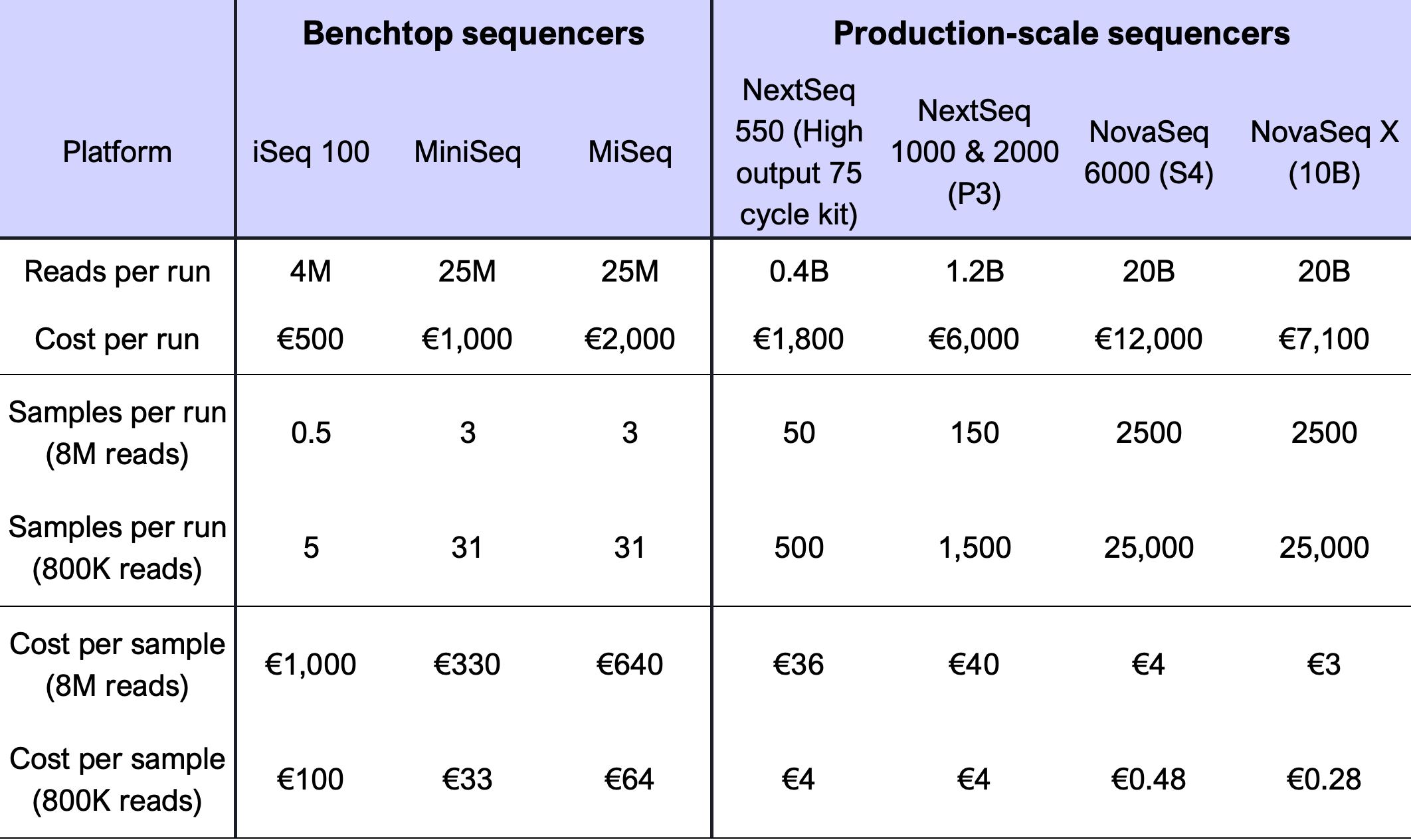
The cost of Bridge Capture™ consists of two parts: sample preparation and sequencing. The sample preparation cost per sample is low regardless of the panel size. The approximated sequencing costs are presented in (Table 1).
The low sample preparation cost is based on the simplicity of the protocol, as well as the small amounts of low-cost reagents (see Publication: Bridge Capture permits cost-efficient, rapid and sensitive molecular precision diagnostics, see question Does Bridge Capture™ have advantages over Molecular Inversion Probes?).
The low sequencing cost is based on the efficient targeting of the sequencing effort, requiring only 800k reads per sample for Tx applications as well as the faithful representation of the read counts in the sample, provided by the linear signal amplification.
Table 1 Bridge Capture™ multiplexing cost on Illumina sequencing platforms using a panel of 282 probes. M = million, B = billion (thousand million).
The Bridge Capture™ workflow is designed to be fast and efficient, taking less than 5 hours to complete with only 5 minutes of hands-on time required, compared to 7 hours on amplicon-based workflows or at least 8 hours on hybrid capture-based workflows. This streamlined workflow allows for rapid sample processing, saving valuable time and resources (See Figure 1).
Once the workflow is complete, the subsequent NGS analysis duration depends on the sequencing technology used.
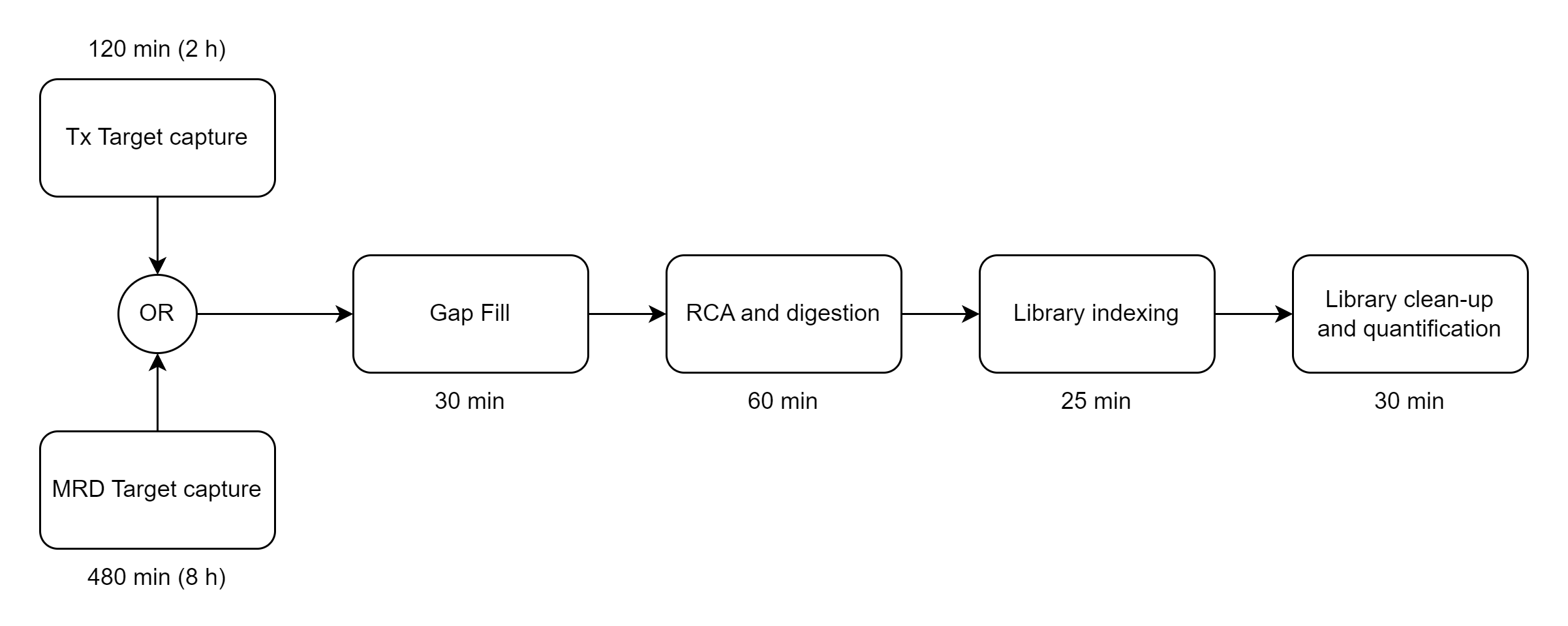
There are multiple optional outputs for Bridge Capture™ and Nicking Loop™.
The key output of both technologies is the circular library (see question How is the Nicking Loop™ conversion & indexing done?), from which the other outputs are derived if needed. The circular library can be achieved in multiple ways with Bridge Capture™ or Nicking Loop™. The circular library can be directly sequenced with a circular DNA -based sequencing technology, such as Complete Genomics’s DNBSEQ , Element’s Aviti and PacBio’s Onso. Prior to sequencing the circular library can also be selectively accessed or unwanted parts of it can be deleted and still retain the original circular library for future use due to the possibility of concurrent circular DNA amplifications.
Optionally, the circular library can be processed further by amplifying it with RCA (see question How does the rolling circle amplification (RCA) work in Bridge Capture™ and Nicking Loop™?), making the newly formed ssDNA concatemer directly adaptable to long-read sequencing platforms, such as Oxford Nanopore Technologies.
From the amplified ssDNA concatemer the individual monomers can be amplified with a limited cycle indexing PCR (see question How does the limited-cycle indexing PCR work in the Bridge Capture™?). The amplicons are traditional linear libraries which can be sequenced with short-read platforms like Illumina and Ion Torrent.
The reproducibility of the read counts reported by Bridge Capture™, determined by replicated sequencing experiments, and indicated as mean-normalized replicate counts, is typically between 0.5x to 2x of the replicate mean (Figure 1).
The sensitivity is reproducible between replicates and was demonstrated experimentally in validation experimentation done on NovaSeq 6000 (see question What parameters affect sensitivity Bridge Capture™?).
The main factors that impact the sensitivity of the Bridge Capture™ are sample quality and total amount of DNA present. It is worth noting that the sensitivity varies slightly between different probes, which is most likely due to the unique binding characteristics determined by the probe and target sequences. In our sensitivity validation experiment, we observed a sensitivity range of 0.03 % to 0.2 % MAF (see Publication: Bridge Capture permits cost-efficient, rapid and sensitive molecular precision diagnostics), which depends on the specific probe. These MAFs were determined through serial dilutions of fragmented tumor biopsy DNA to fragmented gDNA, which were subsequently sequenced on the NovaSeq 6000. We can optimize the performance characteristics of any probes based on customer preferences.
Our clinical testing has not yet been sufficient to establish these numbers. As of 2025-01, we are collecting further sample cohorts to establish numbers for type I (false positive) and type II errors (false negative).
Yes.
Bridge Capture™ workflow has been validated with two local CROs in early 2023 and with one global-scale CRO / precision diagnostic company in mid-2023. The results have been published in medRxiv (see Publication: Bridge Capture permits cost-efficient, rapid and sensitive molecular precision diagnostics).
Yes.
The Bridge Capture™ workflow is highly versatile and can easily be adapted to automated and scalable environments. Recent demonstrations of the workflow were performed with Opentrons OT-2, a popular automation platform for life sciences, where the workflow was completed in a single run.
Bridge Capture™ workflow can be fully automated depending on the chosen automation platform and configuration, encompassing both workflow and library preparation for NGS analysis in the same run. This allows for even greater efficiency and precision in processing large numbers of samples. In addition, the hands-on load time for the automation workflow is brief, taking only approximately 5 minutes to complete.
Yes.
The Nicking Loop™ workflow has been demonstrated with Opentrons OT-2.
No.
Both the Bridge Capture™ and the Nicking Loop™ workflows can be completed using standard molecular laboratory equipment such as pipettes, vortexes, spinners, and PCR instrument. No specialized or uncommon equipment is necessary to successfully carry out the protocol.
Bridge Capture™ is compatible with all NGS platforms on the market. In addition to Illumina platforms, Bridge Capture™ workflow has been successfully completed with Ion Torrent and Oxford Nanopore NGS platforms. Nicking Loop™ has been successfully demonstrated with Illumina platforms. See question What are the outputs of Bridge Capture™ and Nicking Loop™ processes? for an overview of how the technology outputs fit to different sequencing platforms.